



1
BUKU PETUNJUK PRAKTIKUM
SISTEMATIKA MIKROBIA
Oleh:
Dr. Enny Zulaikha, MP.
M. Andry
FAKULTAS BIOLOGI
UNIVERSITAS GADJAH MADA
YOGYAKARTA

2
TAKSONOMI NUMERIK FENETIK
Acara 1 – Klasifikasi Numerik-Fenetik Berdasarkan Data Fenotipik
A. Pengantar
Taksonomi numerik yang juga dikenal dengan sebutan taksonomi Adansonian
(berdasarkan nama ahli sistematika Michael Adanson) didefinisikan sebagai
pengelompokan unit taksonomis ke dalam sejumlah taksa dengan metode numerik
berdasarkan karakteristik yang dimiliki. Taksonomi numerik didasarkan atas lima prinsip
utama, yakni:
1. Taksonomi yang ideal adalah taksonomi yang mengandung informasi terbesar yaitu
yang didasarkan atas sebanyak-banyaknya karakter.
2. Masing-masing karakter diberi nilai yang setara (a priori) dalam mengkonstruksi
takson yang bersifat alami.
3. Tingkat kedekatan antara dua strain/OTU (operational taxonomical unit) merupakan
fungsi similaritas yang dimiliki bersama.
4. Taksa yang berbeda dibentuk atas sifat yang dimiliki.
5. Similaritas tidak bersifat filogenetis melainkan bersifat fenetis.
Tujuan utama taksonomi numerik adalah untuk menghasilkan suatu klasifikasi yang
bersifat teliti, reprodusibel, serta padat informasi. Aplikasi taksonomi numerik dalam
konstruksi klasifikasi biologis memungkinkan terwujudnya sirkumskripsi takson
berdasarkan prinsip yang mantap dan bukan sekedar klasifikasi yang bersifat subjektif
belaka. Teknik klasifikasi meliputi empat tahapan, yaitu:
1. Strain mikrobia (n) yang akan diklasifikasikan dikoleksi lalu ditentukan karakter
fenotipiknya dalam jumlah besar (t) yang mencakup sifat yang tertera pada Tabel 1.1.
Data yang diperoleh disusun dalam suatu matriks n x t.
2. Strain mikrobia diklasifikasikan berdasarkan nilai similaritas atau disimilaritas yang
dihitung dari data n x t.
3. Strain yang mirip akan dimasukkan ke dalam satu kelompok dengan menggunakan
algoritma pengklasteran (clustering algorithm).
4. Kelompok yang dibentuk secara numerik lalu dipelajari dan karakter yang bersifat
membedakan (separating character) dipilih diantara data dalam matriks untuk
selanjutnya digunakan dalam dentifikasi.
B. Cara Kerja
Tahapan kerja dalam klasifikasi numerik-fenetik terdiri dari (1) pemilihan strain uji,
(2) pemilihan jenis pengujian, (3) pencatatan hasil pengujian, (4) data coding, (5) analisis
komputer, dan (6) interpretasi hasil (Priest & Austin, 1993). Namun demikian, dalam
praktikum ini kita akan menggunakan data hasil pengujian dari artikel penelitian yang

3
terdapat pada jurnal ilmiah dalam bentuk tabel n x t. Dengan demikian, kita telah
melewati tahapan kerja 1–3 dan langsung menuju tahapan data coding dan seterusnya.
Tabel 1.1. Kelompok karakter yang digunakan dalam taksonomi numerik bakteri (Priest
& Austin, 1993).
No Kelompok Karakter Jenis Karakter
1
Morfologi kolonial
Bentuk dan ukuran koloni, pigmentasi larut/tidak larut/
fluoresens.
2
Morfologi selular
Sifat pengecatan, bentuk & ukuran sel, motilitas,
ada/tidak nya flagela, ada/tidaknya spora.
3
Sifat pertumbuhan
Bentuk-bentuk pertumbuhan pada medium cair,
kekeruhan, aerobsis/anaerobsis, kebutuhan vitamin,
kemampuan tumbuh pada medium salinitas tinggi.
4
Sifat biokimia
Kemampuan fermentasi/oksidasi karbohidrat,
kehadiran enzim katalase & oksidase, penghasilan
asam, indol, & gas H2S.
5
Resistensi terhadap
antibiotik
Kemampuan untuk tumbuh pada medium yang
mengandung antibiotik.
6
Kemampuan
penggunaan senyawa
kimia sebagai satu-
satunya sumber C
Kemampuan tumbuh pada medium minimal dengan
glukosa/alanin/sitrat sebagai satu-satunya sumber
karbon
7
Sifat serologis
Ada/tidaknya reaksi aglutinasi terhadap antiserum
spesifik
8
Sifat kemotaksonomis
Kehadiran komponen sub-selular tertentu, seperti
peptidoglikan, menakuinon, asam mikolat.
9 Sifat genetik Kandungan G+C pada DNA
10 Phage typing Ada/tidaknya pola phage typing tertentu
1. Penelusuran Sumber Data
Dalam praktikum ini, data untuk klasifikasi numerik-fenetik didapatkan melalui hasil
penelusuran artikel ilmiah. Artikel ilmiah tersebut dapat dicari dari internet melalui
Google Search dengan mengetikan kata-kata kunci terkait klasifikasi numerik fenetik
seperti contohnya “numerical taxonomy of...” atau “numerical classification of...” Salah
satu contoh artikel dapat dilihat pada West et al. (1986) yang mempelajari klasifikasi
spesies anggota genus Vibrio yang diisolasi dari perairan. Data yang terdapat pada artikel
tersebut selanjutnya akan dijadikan contoh pada panduan praktikum ini.
Setelah mendapatkan artikel jurnal, telusuri tabel n x t yang terdapat di dalam artikel
tersebut. Walaupun sering tidak disebut sebagai tabel n x t, namun tabel n x t dapat
terlihat dari cirinya yang mengandung informasi mengenai strain-strain mikrobia (n) serta
jenis karakter yang diujikan (t). Karakter yang terkandung dalam tabel n x t pada
umumnya akan dikelompokan seperti pada Tabel 1.1.

4
2. Konstruksi Tabel n x t
Berdasarkan tabel n x t yang diperoleh dari artikel jurnal penelitian, selanjutnya kita
membuat tabel n x t sendiri. Tabel n x t dapat dibuat dengan program Microsoft Excel
(MS Excel). Pada MS Excel, strain (n) dimasukan sebagai kolom dan karakter pengujian
(t) dimasukan sebagai baris (Gambar 1.1.). Pengkodean pada tabel n x t menggunakan
sistem biner, yakni notasi “1” dan “0”. Notasi “1” diberikan apabila ada kehadiran suatu
sifat dan notasi “0” diberikan untuk ketidakhadiran suatu sifat. Jika dilihat pada Gambar 1
tersebut, kemampuan V. fluvialis untuk dapat tumbuh pada medium dengan kandungan
NaCl 6% menjadikan spesies tersebut mendapatkan notasi “1” pada karakter
“Pertumbuhan 6% NaCl”. Sebaliknya, V. fluvialis tidak dapat tumbuh pada medium
dengan kandungan NaCl 0% sehingga membuatnya mendapatkan notasi “0” untuk sifat
tersebut. Dalam laporan tertulis atau publikasi ilmiah, penggunaan notasi “1” dan “0”
pada n x t sebagai indikasi kehadiran dan ketidakhadiran suatu sifat pada suatu strain
terkesan kurang lazim. Pada umumnya, notasi “+” dan “–“ digunakan sebagai pengganti
“1” dan “0”. Namun demikian, notasi “1” dan “0” pada n x t dimaksudkan untuk
kepentingan analisis menggunakan program komputer, karena program tersebut tidak
dapat mengenali notasi “+” dan “–“.
Gambar 1.1. Contoh tabel n x t
Perlu diketahui bahwa pemilihan spesies dalam konstruksi tabel n x t sebaiknya
mencakup type strain untuk spesies tersebut. Dengan kata lain, sangat disarankan apabila
data yang digunakan juga memiliki hasil pengujian untuk type strain. Suatu type strain
merupakan strain yang mewakili suatu spesies atau dengan kata lain type strain
merupakan pemilik nama spesies. Kehadiran suatu type strain dalam proses konstruksi
dendrogram berfungsi sebagai acuan bagi strain-strain lainnya yang memiliki kesamaan
nama spesies dengan type strain, sehingga dapat lebih meyakinkan posisi strain-strain
tersebut dalam suatu dendrogram.
3. Konstruksi Dendrogram Dengan Program MVSP 3.1 (Kovach, 2007)
Konstruksi dendrogram dengan MVSP 3.1 membutuhkan file input dalam format
.mvs. File tersebut dapat dihasilkan menggunakan program Programmer’s File Editor
(PFE) atau Notepad. Kedua program tersebut memiliki kalimat perintah yang sama,
sehingga dalam penjelasan ini hanya akan diterangkan mengenai satu program saja yakni
PFE. Pada program PFE, buka file baru “New” dan akan tampil sebuah layar. Pada layar
tersebut ketik *L <jumlah karakter> <jumlah strain>. Sebagai contoh,

5
apabila kita ingin mengkonstruksi dendrogram 10 spesies dengan 50 karakter, maka kita
akan menuliskan *L 50 10 pada baris pertama layar.
Setelah itu, copy tabel n x t dari MS Excel ke dalam layar PFE. Namun sebelum itu,
tabel n x t harus sedikit dimodifikasi agar dapat terbaca oleh program PFE dan MVSP.
Modifikasi yang dilakukan meliputi penambahan baris dibawah nama OTU dan kolom
disamping jenis karakter (Gambar 2). Penambahan ini dilakukan karena program PFE dan
juga MVSP akan memperlakukan spasi sebagai pemisah antar dua kategori. Apabila kita
tidak menambahkan simbol pengganti karakter atau OTU, maka sebagai contoh “V.
fluvialis” akan dianggap sebagai dua kategori yakni “V.” dan “fluvialis”. Setelah
modifikasi selesai dilakukan, maka tabel n x t selanjutnya di-copy ke program PFE. Perlu
diketahui bahwa hanya character states dan simbol karakter yang di-copy-kan ke PFE
(kotak abu muda dan abu tua pada Gambar 1.2.).
(a)
(b)
Gambar 1.2. (a) Tabel n x t awal, dan (b) modifikasi berupa penambahan baris simbol
strain [a, b, c] dan kolom simbol karakter [1, 2, 3, 4, 5].
Hasil copy akan terlihat seperti berikut:
*L 5 3
a b c
1 0 0 0
2 1 1 1
3 1 1 0
4 0 0 0
5 0 0 0
Simpan file tersebut dalam format .mvs dengan memberikan tambahan “.mvs” pada
akhir nama file dalam menu save. File .mvs tersebut selanjutnya dapat dibuka dengan
program MVSP 3.1. Selanjutnya, masuk ke program MVSP 3.1 dengan membuka
aplikasi mvspw. Pada MVSP, klik File → Open dan pilih file .mvs yang tersimpan.

6
Ketika terbuka, MVSP akan menampilkan sebuah layar tambahan mengenai jumlah
variabel dan jumlah sampel dari file .mvs yang kita masukan. Pada program MVSP, kita
dapat mengubah kembali nama OTU/sampel yang sebelumnya telah diubah menjadi
simbol [a, b, c] dengan menu Data → Edit Data. Pengubahan data pada menu
tersebut dapat menerima spasi, namun tidak dapat mencetak miring sebagaimana yang
seringkali dilakukan terhadap nama suatu spesies. Setelah semua simbol OTU diubah
menjadi nama spesiesnya, kita dapat menutup layar Edit Data tersebut dan memulai
analisis klaster.
Analisis klaster dilakukan melalui menu Analyses → Cluster Analysis.
Pada layar Cluster Analysis, kita dapat menentukan jenis perhitungan indeks
similaritas dan analisis klaster yang diinginkan. Dalam praktikum ini kita akan mencoba
menggunakan dua jenis perhitungan indeks similaritas (SSM dan SJ) dan metode
pengklasteran average linkage / UPGMA. Pada metode pengklasteran, kita juga
dapat memilih single linkage / nearest neighbour atau complete linkage /
farthest neighbour. Setelah selesai memilih perhitungan indeks similaritas dan
metode pengklasteran pada submenu Options, kita dapat memilih output analisis pada
menu Advanced. Pada submenu Advanced, kita dapat menentukan penampilan hasil
analisis pada kolom result to display. Nyalakan pilihan
Similarity/distance matrix, Clustering report, dan Text
dendrogram. Setelah semuanya selesai, maka klik OK.
Hasil analisis akan memunculkan dua layar, yakni layar pertama yang berisikan
matriks similaritas dan analisis klaster serta layar kedua yang menampilkan dendrogram.
Untuk memudahkan analisis selanjutnya, disarankan untuk meng-copy seluruh isi layar
pertama yang menyerupai layar Excel ke program MS Excel. Gambar dendrogram dapat
dijadikan file gambar (image file) dengan File → Export.
4. Analisis Korelasi-Kofenetik
Analisis korelasi-kofenetik dilakukan untuk melihat tingkat akurasi suatu
dendrogram yang merepresentasikan matriks similaritas antar OTU. Analisis ini
membandingkan dua jenis matriks similaritas, yakni matriks similaritas awal (unsorted)
dan matriks similaritas yang diturunkan dari dendrogram (sorted). Analisis ini dapat
dilakukan dengan program MS Excel. Sebelum mengisikan nilai-nilai dari kedua matriks
similaritas, kita perlu membuat pasangan OTU terlebih dahulu. Jumlah kemungkinan
pasangan OTU yang dapat dibentuk dari n-OTU dijabarkan dengan rumus kombinasi:
Pada contoh diatas, kita dapat menghasilkan 3 jenis kemungkinan pasangan dari total 3
OTU, yakni [a,b], [a,c], dan [b,c]. Setelah semua kemungkinan pasangan dituliskan,
masukan nilai untuk kolom unsorted, yakni nilai similaritas dari matriks similaritas awal

7
dan kemudian nilai untuk kolom sorted yang berasal dari dendrogram / analisis klaster.
Dari hasil MVSP kita memperoleh matriks similaritas:
V. fluvialis V. furnisii V. natriegens
V. fluvialis 1.000
V. furnisii 1.000 1.000
V. natriegens 0.500 0.500 1.000
dan analisis klaster:
Objects
Node Group 1 Group 2 Simil. in group
1 V. fluvialis V. furnisii 1.000 2
2 Node 1 V. natriegens 0.500 3
Berdasarkan matriks similaritas tersebut, kita memasukan nilai unsorted. Dengan
demikian, pasangan [a,b] yang dalam hal ini [V. fluvialis, V. furnisii] memiliki nilai 1.000.
Hal yang sama untuk pasangan [a, b] juga sama pada analisis klaster, sehingga kolom
sorted untuk pasangan [a,b] bernilai 1.000. Hal yang perlu diperhatikan, khususnya pada
tabel analisis klaster adalah pada nomor 2, yakni penggabungan Node 1 dengan V.
natriegens. Dalam hal ini, masing-masing komponen dari Node 1, yakni a dan b akan
berpasangan dengan V. natriegens ([c]) pada nilai 0.500. Hal ini berarti kombinasi [a,b]
dengan [c] akan menghasilkan [a,c] dan [b,c] yang keduanya memiliki nilai similaritas
0,500.
Setelah semua komponen nilai dimasukan, maka akan terbentuk tabel seperti:
OTU Unsorted Sorted
ab 1 1
ac 0.5 0.5
bc 0.5 0.5
...
Dari tabel tersebut, kita dapat menghitung nilai korelasi menggunakan fungsi CORREL.
Indeks korelasi yang diterima adalah ≥0,7 atau ≥70%. Semakin rendah nilai korelasi
berarti dendrogram yang terbentuk semakin tidak mewakili matriks similaritas yang
menjadi dasar konstruksinya.
5. Penampilan Hasil
Data dan hasil yang diperoleh dari praktikum ini meliputi: (1) tabel n x t, (2) matriks
similaritas, (3) analisis klaster, (4) dendrogram, dan (5) analisis korelasi-kofenetik. Perlu
diingat bahwa setiap entry pada tabel n x t yang sebelumnya dimasukan dengan notasi “1”
dan “0” perlu diganti dengan notasi “+” dan “–“ untuk pelaporan hasil. Perubahan ini
dapat dilakukan pada MS Excel menggunakan fungsi Find/Replace (ctrl+F). Seluruh data
dan hasil dimuat dalam bab hasil pada laporan praktikum.

8
Acara 2 – Klasifikasi Numerik-Fenetik Berdasarkan Profil Sidik Jari Molekular
A. Pengantar
Selain data fenotipik, data berupa profil sidik jari molekular (molecular fingerprints)
juga dapat digunakan dalam studi taksonomi mikrobia. Profil yang digunakan dapat
berasal dari DNA, RNA, atau protein. Studi klasifikasi menggunakan profil sidik jari
molekular termasuk ke dalam studi kemosistematik bersama dengan analisis komponen
selular lainnya (Priest & Austin, 1993). Teknik yang paling umum digunakan dalam studi
klasifikasi berdasarkan profil sidik jari molekular adalah restriction fragment length
polymorphisms (RFLP). Prinsip dasar dari penggunaan RFLP adalah adanya kesamaan
situs restriksi antara satu mikroorganisme dengan mikroorganisme lainnya yang dapat
dikenali oleh enzim restriksi endonuklease tertentu. Hal ini berarti bahwa distribusi situs-
situs restriksi tersebut dapat memberi gambaran mengenai kemiripan antara satu
mikroorganisme dengan lainnya secara molekular.
B. Cara Kerja
Seperti halnya pada praktikum acara 1, data yang dipakai dalam praktikum acara 2
ini diperoleh dari artikel penelitian yang terdapat pada jurnal ilmiah dalam bentuk
elektroforegram (foto elektroforesis).
1. Penelusuran Sumber Data
Data yang digunakan dalam praktikum ini berupa elektroforegram sidik jari
(fingerprint) molekular. Data fingerprint tersebut dapat berupa hasil ribotyping, RFLP,
RAPD, dan lainnya. Elektroforegram dari suatu artikel jurnal dapat di-crop menggunakan
program PDF reader seperti Adobe Reader melalui menu Tools → Select & Zoom
→ Snapshot Tool. Buat file gambar (image file) format JPEG atau PNG dari
elektroforegram tersebut dengan memblok elektroforegram yang diinginkan dan copy
(ctrl+C) ke program Paint.
2. Pembuatan Diagrammatic Representative
Diagrammatic representative merupakan sebuah gambaran skematis yang mewakili
elektroforegram yang dianalisis. Penggunaan diagrammatic representative ini barfungsi
untuk mempertegas kehadiran atau ketidakhadiran suatu band dalam elektroforegram.
Kejelasan mengenai hadir atau tidaknya sebuah band merupakan hal yang penting karena
band tersebut diasumsikan sebagai karakter dalam data coding.
Pembuatan diagrammatic representative dapat dilakukan dengan program photo
editor seperti Corel Draw, Adobe Photoshop, atau Paint Shop Pro. Secara umum,
tahapan pembuatannya meliputi input gambar, penambahan layer transparan, dan
menggambarkan suatu garis pada layer transparan tersebut yang berpandu pada band
elektroforegram dibawahnya. Dalam praktikum ini program Paint Shop Pro (PSP) akan
digunakan untuk pembuatan diagrammatic representative ini. Pertama, buka program
PSP dan masukan file gambar elektroforegram yang telah dibuat sebelumnya melalui

9
menu File → Open. Setelah gambar elektroforegram muncul, akan terdapat suatu kotak
bertuliskan Layer pada sisi sebelah kanan. Apabila kotak ini tidak muncul, kita dapat
memunculkannya melalui ikon Toggle Layer Palette pada sisi atas, ikon kedua
dari kanan. Pada layer palette tersebut, gambar yang kita masukan sebelumnya
dikategorikan sebagai Background. Kita dapat menambahkan layer baru dengan
menekan ikon Add New Layer yang terletak pada pojok kiri bawah layer
palette. Sebuah layer baru yang muncul akan diberi nama Layer 1 secara otomatis
dan terletak di atas layer Background. Klik ikon Layer 1 untuk mengaktifkannya
dan Pada Layer 1 inilah kita akan membuat diagrammatic representative dari
elektroforegram.
Setelah memilih Layer 1, aktifkan control palette dengan menekan ikon Toggle
Control Palette yang terletak 3 ikon di sebelah kiri Toggle Layer Palette.
Menu control palette berfungsi untuk mengatur jenis, ketebalan, bentuk, dan ukuran
brush tip yang akan digunakan untuk mewarnai Layer 1 berdasdarkan cetakan yang
ada pada elektroforegram. Sebelum mengatur apapun yang ada pada control palette,
aktifkan Paint Brushes yang terletak pada tool palette (menu di pojok kiri).
Pemilihan warna dapat dilakukan pada color palette (menu di pojok kanan). Kedua menu
tersebut masing-masing dapat diaktifkan melalui Toggle Tool Palette dan
Toggle Color Palette. Menu Control Palette terdiri atas dua submenu,
yakni Tool Controls dan Brush Tip. Untuk sementara, abaikan dahulu submenu
Tool Controls dan masuk ke submenu Brush Tip. Pada submenu Brush Tip
terdapat pilihan Shape, Size, Opacity, Hardness, Density, dan Step yang
masing-masing berfungsi untuk mengatur jenis kuas, ukuran kuas, ketebalan warna,
ketegasan bentuk kuas, kepadatan warna, dan gradasi kuas. Hasil pengaturan enam
parameter ini langsung ditampilkan pada layar di kiri atas submenu Brush Tip.
Sebagai contoh, pengaturan Brush Tip yang digunakan meliputi Shape:
Horizontal, Opacity: 25, Hardness: 50, Density: 100, dan Step: 1.
Size yang digunakan bergantung pada ukuran image file yang digunakan. Sebelum
mulai membuat diagrammatic representative, sebaiknya kita mengujikan hasil pengaturan
pada submenu Brush Tip untuk melihat kecocokannya langsung pada Layer 1.
Hasil pengujian kemudian dapat dihapus dengan mengaktifkan ikon Eraser pada tool
palette. Bentuk dan ukuran Eraser juga diatur pada menu Control Palette yang
sama dengan Brush Tip. Hasil penggambaran dapat dilihat pada Gambar 2.1.
Setelah penggambaran selesai dilakukan, simpan file tersebut (PSP akan secara
otomatis menyimpannya dalam format .psp). File .psp ini juga dapat dibuka dengan
program image viewer seperti ACDSee. Diagrammatic representative yang ditampilkan
adalah hasil penggambaran yang dilakukan tanpa menyertakan background foto
elektroforegram.

10
Gambar 2.1. (a) Elektroforegram awal yang dijadikan latar belakang/background dan (b)
hasil penggambaran yang berpandu pada latar belakang tersebut.
Dengan demikian, kita dapat menghilangkan foto elektroforegram dengan meng-klik
ikon Background pada Layer Palette dan kemudian Delete. Hasilnya hanya
akan tersisa pita-pita berwarna hasil penggambaran sebelumnya (Gambar 2.2). Simpan
kembali file ini dalam format .jpg (gunakan menu Save As) dengan nama lain agar
tidak mengganti file sebelumnya.
Gambar 2.2. (a) Elektroforegram dan (b) diagrammatic representative yang dihasilkan
dari elektroforegram tersebut.
3. Data Coding dan Analisis
Diagrammatic representative yang dihasilkan merupakan sumber untuk data coding.
Dalam hal ini, setiap pita yang tergambarkan pada diagrammatic representative
merupakan karakter, sehingga total seluruh pita dari seluruh strain mencerminkan jumlah
karakter (t) pada tabel n x t. Konversi diagrammatic representative menjadi tabel n x t
dapat dilakukan secara intuitif dengan melihat kesejajaran pita antar strain dan
menggolongkan pita-pita yang sejajar sebagai satu karakter (Gambar 2.3). Namun
demikian, apabila jumlah pita terlalu banyak maka diperlukan garis bantu untuk melihat
kesejajaran antar pita. Garis bantu tersebut dapat dibuat pada program Paint. Buka file
gambar diagrammatic representative dalam format .jpg dengan program Paint dan klik
ikon Line yang terletak pada bagian atas. Setelah itu buat garis dari ujung kiri ke ujung
kanan gambar dimulai dari pita teratas. Pita-pita yang bersinggungan dengan garis
tersebut dinyatakan sejajar dan menempati karakter yang sama.

11
Gambar 2.3. Konversi diagrammatic representative menjadi tabel n x t.
Tabel n x t yang telah dibuat selanjutnya dianalisis secara numerik-fenetik sama seperti
pada analisis data fenotipik. Analisis dilakukan menggunakan program MVSP 3.1
(Kovach, 2007) dengan indeks similaritas SSM dan SJ serta algoritme pengklasteran
UPGMA.
4. Interpretasi Hasil
Hasil analisis numerik-fenetik data fingerprint molekular ini sekilas terlihat sama
dengan data fenotipik, yakni terdiri atas matriks similaritas, analisis klaster, dan
dendrogram. Akan tetapi, perlu diperhatikan bahwa kaidah yang diterapkan pada data
fenotipik tidak harus diterapkan pada data fingerprint ini. Salah satu syarat seperti
digunakannya minimal 50 karakter pada data fenotipik tidak perlu diterapkan pada data
fingerprint. Hal ini didasarkan pada fakta bahwa profil fingerprint yang ada dipandang
hanya sebagai salah satu karakter saja dan tabel n x t yang dikonstruksi merupakan
turunan dari satu karakter tersebut.

12
TAKSONOMI FILOGENETIK
Acara 3 – Klasifikasi Molekular Filogenetik Berdasarkan Gen 16S rRNA
A. Pengantar
Klasifikasi molekular filogenetik merupakan klasifikasi yang disusun dengan
mempertimbangkan jalur evolusi setiap organisme yang dikaji. Hal ini berbeda dengan
klasifikasi fenetik yang hanya melihat hubungan antar organisme berdasarkan karakter
yang ada pada saat ini (Priest & Austin, 1993). Dalam prosesnya, klasifikasi molekular
filogenetik menggunakan data berupa urutan (sequence) nukleotida pada DNA atau asam
amino pada protein. Sequence nukleotida yang digunakan dalam klasifikasi molekular
filogenetik harus merupakan sequence yang diwariskan langsung oleh nenek moyang
(homolog) serta memiliki kesamaan sejarah evolusi. Sebuah sequence dapat disebut
sebagai marker molekular apabila memenuhi persyaratan berupa: (1) terdistribusi pada
seluruh organisme, (2) memiliki kesetaraan fungsi pada seluruh organisme, dan (3)
memegang peranan vital bagi kehidupan organisme. Hal ini menjadikan marker
molekular merupakan sequence yang tepat untuk digunakan dalam studi klasifikasi
filogenetik.
Gen 16S rRNA merupakan salah satu contoh marker molekular karena terdapat pada
organisme baik yang berada pada domain Bacteria, Archaea, serta Eukarya. Saat ini,
informasi mengenai sequence gen 16S rRNA sudah sangat banyak tersedia pada database
internasional dan juga sudah dijadikan standar penetapan suatu spesies baru dalam studi
taksonomi. Pada praktikum ini kita akan mencoba mengklasifikasikan sejumlah bakteri
dengan cara merekonstruksi pohon filogenetik berdasarkan sequence gen 16S RNA yang
dimiliki.
B. Cara Kerja
3.1.1. Persiapan Data
Data yang digunakan dalam klasifikasi berbasis molekular filogenetik adalah berupa
urutan (sequence) nukleotida atau asam amino. Kedua jenis sequence ini umumnya dapat
diperoleh dari hasil sequencing DNA/protein maupun dari sequence database
internasional yang tersedia di internet. Untuk kemudahan dalam praktikum ini, kita akan
menggunakan cara kedua, yakni mengunduh sequence nukleotida/asam amino dari
internet. Saat ini sudah banyak sekali situs yang memfasilitasi hal tersebut. Situs
GenBank, DDBJ, serta EMBL merupakan situs dimana pencarian sequence
nukleotida/asam amino umumnya dilakukan. Dalam acara praktikum ini, kita akan
mencoba mengunduh data melalui GenBank (http://www.ncbi.nlm.nih.gov/Genbank/)
dan untuk selanjutnya sequence DNA akan digunakan sebagai contoh persiapan data.
Pada GenBank kita dapat mencari sequence DNA berdasarkan nama gen, spesies
pemilik gen, atau accession number pada kolom search yang tersedia. Accession number

13
merupakan penanda/identitas dari setiap sequence DNA yang telah disimpan pada situs
tersebut. Pada umumnya accession number tertulis pada artikel publikasi ilmiah yang
penelitinya telah menyumbangkan hasil sequencing-nya ke database yang ada. Pencarian
pada menu search di GenBank terdiri dari banyak menu sub-pencarian untuk
memudahkan penelusuran data, namun dalam praktikum ini kita akan lebih fokus pada
pencarian dengan menu Nucleotide, Protein, dan Genome. Sebagai contoh, apabila kita
ingin melakukan pencarian gen 16S rRNA bakteri Escherichia coli maka kita dapat
melakukan salah satu dari hal berikut:
1. Melakukan penelusuran pustaka, misalnya pada artikel jurnal untuk mendapatkan
accession number. Sebagai contoh, Escherichia coli ATCC 11775T pada Bergey’s
Manual of Systematic Bacteriology (Brenner et al., 2005) memiliki accession number
X80725. Accession Number ini dapat diketikan pada kolom search nucleotide.
Cara ini merupakan pencarian yang cukup spesifik karena setiap sequence dari setiap
jenis organisme memiliki accession number tersendiri.
2. Melakukan penelusuran pada situs List of Prokaryotic names with Standing in
Nomenclature (LPSN, http://www.bacterio.cict.fr/). Penelusuran melalui situs ini
khusus untuk pencarian sequence bakteri dan archaea saja. Informasi yang diberikan
mengenai suatu genus/spesies meliputi type species, type strain, tahun ditemukan, dan
publikasi (valid/efektif) terkait genus/spesies tersebut.
3. Melakukan penelusuran pada kolom search nucleotide di GenBank dengan
mengetik kata kunci, seperti “Escherichia coli 16S rRNA”. Cara ini kurang spesifik
karena akan menghasilkan sejumlah daftar yang berisikan berbagai macam spesies
bakteri yang mengandung sequence gen 16S rRNA dan kita harus mencarinya satu per
satu.
4. Melakukan penelusuran pada kolom search genome di GenBank dan kemudian
mengetik nama spesies yang kita inginkan. Umumnya hasil penelusuran meliputi
sejumlah daftar complete genome dari spesies yang dicari. Kita dapat mencari gen
yang diinginkan dalam daftar complete genome tersebut. Cara ini cukup memakan
waktu, namun kita dapat menelusuri berbagai jenis gen dari spesies tersebut.
Contoh Hasil pencarian dengan accession number X80725 akan ditampilkan sebagai
berikut:
E.coli (ATCC 11775T) gene for 16S rRNA
GenBank: X80725.1
FASTA Graphics
LOCUS X80725 1450 bp DNA
linear BCT 29-MAR-1996
DEFINITION E.coli (ATCC 11775T) gene for 16S rRNA.
ACCESSION X80725
VERSION X80725.1 GI:1240022

14
KEYWORDS 16S ribosomal RNA; 16S rRNA gene; 16S small
subunit ribosomal RNA.
SOURCE Escherichia coli DSM 30083
ORGANISM Escherichia coli DSM 30083
Bacteria; Proteobacteria; Gammaproteobacteria;
Enterobacteriales;
Enterobacteriaceae; Escherichia.
REFERENCE 1
AUTHORS Cilia,V., Lafay,B. and Christen,R.
TITLE Sequence heterogeneities among 16S ribosomal RNA
sequences, and their effect on phylogenetic analyses at the
species level
JOURNAL Mol. Biol. Evol. 13 (3), 451-461 (1996)
PUBMED 8742634
REFERENCE 2 (bases 1 to 1450)
AUTHORS Lafay,B.
TITLE Direct Submission
JOURNAL Submitted (26-JUL-1994) B. Lafay, CNRS &
Universite Paris 6,Station Zoologique, Observatoire
Oceanologique, Villefranche Sur
Mer, 06230, FRANCE
FEATURES Location/Qualifiers
source 1..1450
/organism="Escherichia coli DSM 30083"
/mol_type="genomic DNA"
/strain="ATCC 11775T"
/db_xref="taxon:866789"
gene 1..1450
/gene="16S rRNA"
rRNA 1..1450
/gene="16S rRNA"
/product="16S ribosomal RNA"
ORIGIN
1 agtttgatca tggctcagat tgaacgctgg cggcaggcct aacacatgca
agtcgaacgg
61 taacaggaag cagcttgctg ctttgctgac gagtggcgga cgggtgagta
atgtctggga
121 aactgcctga tggaggggga taactactgg aaacggtagc taataccgca
taacgtcgca
181 agcacaaaga gggggacctt agggcctctt gccatcggat gtgcccagat
gggatta...

15
Tulisan diatas mengandung informasi mengenai jenis sequence, asal sequence, produk
yang dihasilkan, dan sequence itu sendiri. Dalam contoh ini jenis sequence adalah gen
16S rRNA yang berasal dari Escherichia coli ATCC 11775T. Gen ini akan menghasilkan
produk berupa RNA ribosomal (rRNA). Informasi mengenai hasil pengkodean dari
sequence dapat dilihat pada FEATURES. Sequence dari DNA itu sendiri yang dituliskan
per 10 nukleotida pada kolom ORIGIN. Sequence ini selanjutnya dapat diunduh dalam
bentuk FASTA dengan cara meng-klik link FASTA pada baris ketiga, dan akan muncul:
E.coli (ATCC 11775T) gene for 16S rRNA
GenBank: X80725.1
GenBank Graphics
>gi|1240022|emb|X80725.1| E.coli (ATCC 11775T) gene for 16S
rRNA
AGTTTGATCATGGCTCAGATTGAACGCTGGCGGCAGGCCTAACACATGCAAGTCGAACGG
TAACAGGAAGCAGCTTGCTGCTTTGCTGACGAGTGGCGGACGGGTGAGTAATGTCTGGGA
AACTGCCTGATGGAGGGGGATAACTACTGGAAACGGTAGCTAATACCGCATAACGTCGCA
AGCACAAAGAGGGGGACCTTAGGGCCTCTTGCCATCGGATGTGCCCAGATGGGATTA...
Kedua jenis data ini (informasi sequence dan sequence FASTA) dikompilasi ke masing-
masing arsip untuk membuat database sendiri.
1. Sequence Alignment dengan Program ClustalX 2.1 (Larkin et al., 2001)
Alignment bertujuan untuk menata sequence agar satu sama lain diletakkan sesuai
dengan posisi homologi antar sequence. Hanya berdasarkan alignment inilah kita dapat
membandingkan antar sequence gen 16S rRNA dari masing-masing strain mikrobia yang
akan diklasifikasikan. Alignment menggunakan program ClustalX dilakukan dengan
mempersiapkan data sequence dalam format FASTA. Dataset sejumlah sequence yang
telah didapatkan dikumpulkan terlebih dahulu ke dalam 1 file Notepad dengan awalan
dari setiap sequence diberikan tanda “>”. Contohnya dapat dilihat pada sequence berikut:
>Allochromatium vinosum DSM 180
agagtttgatcctggctcagattgaacgctggcggcatgcctaacacatgcaa...
>Chlorobium luteolum DSM 273
aggaaagcggcttcggccgggagtacttggcgcaagggtgagtaaggcatagg...
>Chloroflexus aurantiacus J-10-fl
aaaggaggtgatccagccgcaccttccggtacggctaccttgttacgacttcg...
Penamaan dalam pembuatan dataset sequence ini juga perlu diperhatikan. Disarankan
untuk menyingkat nama dari setiap sequence yang ada karena program ClustalX akan
secara otomatis memotong karakter nama apabila melebihi 30 karakter. Selain itu, hasil
alignment ClustalX yang akan digunakan dalam program Phylip (.phy) juga akan secara
otomatis memotong karakter nama apabila melebihi 10 karakter. Hal ini akan
membingungkan apabila pembeda antar nama sequence satu dengan lainnya terletak pada
16
posisi karakter >10, karena ketika dipotong akan menghasilkan nama yang sama antar
sequence satu dengan lainnya. Dengan demikian, sebaiknya kita membuat daftar baru
(dengan MS Word atau Notepad) yang berisi rincian nama sequence beserta
singkatannya. Singkatan nama ini yang akan kita gunakan dalam dataset yang akan
diproses dalam ClustalX. Contoh dataset diatas setelah namanya disingkat akan menjadi:
>AvinDSM180
agagtttgatcctggctcagattgaacgctggcggcatgcctaacacatgcaa...
>ClutDSM273
aggaaagcggcttcggccgggagtacttggcgcaagggtgagtaaggcatagg...
>ChaurJ-10-fl
aaaggaggtgatccagccgcaccttccggtacggctaccttgttacgacttcg...
Perlu diperhatikan bahwa pemberian nama tidak boleh mengandung spasi. Penyingkatan
nama sequence hingga satu karakter (A-Z) sebaiknya tidak dilakukan karena akan
membuat program ClustalX salah mengenali karakter nama menjadi salah satu komponen
sequence nukleotida atau asam amino. Jadi, contoh seperti:
>A
caaaatggagagtttgatcctggctcaggatgaacgctggcggcgtgcttaac...
tidak boleh dilakukan karena nama “A” akan disalahartikan sebagai nukleotida oleh
ClustalX. Dataset yang telah siap selanjutnya disimpan dan selanjutnya dimasukan ke
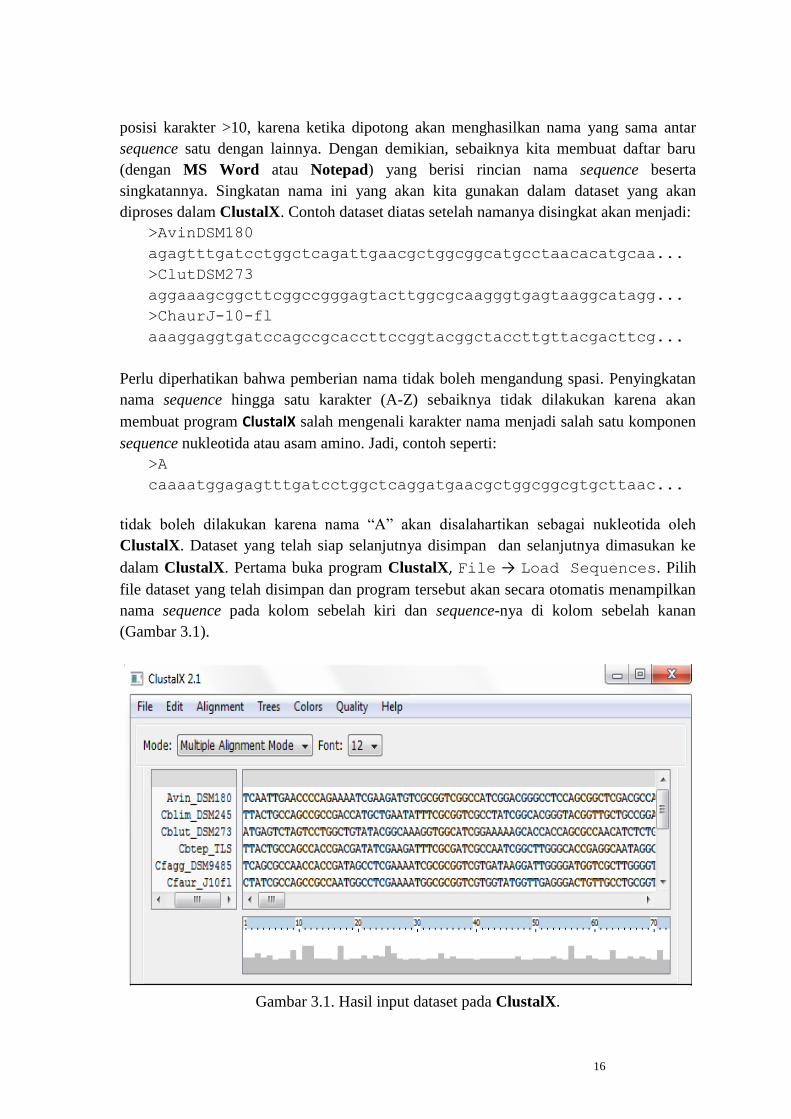
dalam ClustalX. Pertama buka program ClustalX, File → Load Sequences. Pilih
file dataset yang telah disimpan dan program tersebut akan secara otomatis menampilkan
nama sequence pada kolom sebelah kiri dan sequence-nya di kolom sebelah kanan
(Gambar 3.1).
Gambar 3.1. Hasil input dataset pada ClustalX.

17
Sebelum melakukan alignment, kita harus terlebih dahulu mengatur file output yang akan
dihasilkan. Program ClustalX dapat menghasilkan beberapa macam format file dari satu
jenis dataset, yakni: CLUSTAL, GCG/MSF, GDE, NBRF/PIR, NEXUS dan
PHYLIP. Masing-masing file output ini mempunyai kegunaan masing-masing karena
diperlukan oleh program-program analisis lainnya. Dalam praktikum ini kita hanya akan
menggunakan format CLUSTAL (.aln), GDE (.gde), dan PHYLIP (.phy). Pada program
ClustalX, pilih Alignment → Output Format Options → klik GDE FORMAT,
dan PHYLIP FORMAT → CLOSE. Setelah itu pilih Alignment → Do Complete
Alignment.
3.1.2. Rekonstruksi Pohon Filogenetik dengan PHYLIP v3.6.9 (Felsenstein, 2005)
Rekonstruksi dengan program ini membutuhkan file input dalam format .phy yang
dapat dihasilkan oleh program ClustalX atau MEGA. Program Phylip memiliki
serangkaian aplikasi executeable yang dapat menganalisis data sequence dengan berbagai
algoritme. Aplikasi yang terdapat pada Phylip antara lain:
clique dnamlk drawgram neighbor restml
concense dnamove drawtree pars retree
contml dnapars factor penny seqboot
contrast dnapenny fitch proml treedist
dnacomp dollop gendist promlk
dnadist dolmove main protdist
dnainvar dolpenny mix protpars
dnaml draw move restdist
Rekonstruksi pohon filogenetik pada Phylip dapat dilakukan dengan aplikasi
neighbor, fitch, dnapars, dan dnaml untuk sequence nukleotida. Aplikasi
neighbor juga dapat digunakan untuk merekonstruksi pohon dengan sequence asam
amino, namun sebelumnya harus menggunakan input data yang dihasilkan dari aplikasi
protdist. Aplikasi protml dan protpars memiliki prinsip analisis yang sama
dengan dnaml dan dnapars, namun menggunakan sequence asam amino sebagai data
inputnya.
Copy file format .phy ke dalam folder exe yang terdapat di dalam folder Phylip dan
kemudian ganti nama file tersebut menjadi infile tanpa menggunakan
extension/format apapun. Folder exe terkadang sudah mengandung file dengan nama
infile yang disebabkan oleh analisis data yang pernah dilakukan sebelumnya. Apabila
kita menjumpai file infile tersebut, hapus terlebih dahulu dan kemudian baru
mengganti nama file .phy menjadi infile. Selanjutnya, buka salah satu dari aplikasi
yang tersedia dalam folder exe untuk menganalisis infile, dalam hal ini kita akan
menggunakan aplikasi neighbor, yakni rekonstruksi dengan algoritme Neighbor-
Joining.

18
Perlu diketahui bahwa rekonstruksi pohon filogenetik yang menggunakan data
distance matrix berbasis clustering seperti UPGMA, Neighbor-Joining, dan algoritme
Fitch-Margoliash menggunakan data berupa matriks p-distance yang dihasilkan oleh
aplikasi dnadist untuk sequence DNA atau protdist untuk sequence asam amino.
Dengan demikian, infile perlu dianalisis terlebih dahulu dengan dnadist,
kemudian outfile hasil analisis dnadist diubah lagi (rename) menjadi infile
untuk dapat dianalisis dengan neighbor. Setelah namanya diubah, buka aplikasi
neighbor dan akan tampil menu berupa:
Neighbor-Joining/UPGMA method version 3.69
Settings for this run:
N Neighbor-joining or UPGMA tree? Neighbor-joining
O Outgroup root? No, use as outgroup species 1
L Lower-triangular data matrix? No
R Upper-triangular data matrix? No
S Subreplicates? No
J Randomize input order of species? No. Use input order
M Analyze multiple data sets? No
0 Terminal type (IBM PC, ANSI, none)? ANSI
1 Print out the data at start of run No
2 Print indications of progress of run Yes
3 Print out tree Yes
4 Write out trees onto tree file? Yes
Y to accept these or type the letter for one to change
Tekan huruf yang bersangkutan untuk mengubah pengaturan yang diinginkan. Setelah
selesai, tekan Y dan Enter. File hasil analisis akan dimuat ke dalam outfile dan
gambar pohon dimuat pada outtree. File outfile dapat dibuka dengan Notepad dan
dapat di-copy ke dalam MS Excel untuk memudahkan pembacaan, sedangkan file
outtree dapat dibuka dengan program Treeview (Page, 1996).
Rekonstruksi pohon filogenetik dengan algoritme berbasis character-based seperti
maximum parsimony dan maximum likelihood menggunakan file .phy sebagai infile
dan tidak perlu dibuat matriks p-distance. Dengan demikian kita dapat langsung
menjalankan program dnapars untuk analisis parsimony atau dnaml untuk analisis
likelihood langsung terhadap infile.
3.1.3 Pembuatan Matriks Similaritas DNA dengan Phydit (Chun, 1995)
Program Phydit menggunakan data input berupa hasil alignment dengan format .gde
yang dapat dihasilkan dengan program ClustalX. Salah satu analisis yang dilakukan oleh
Phydit adalah penghasilan matriks similaritas nukleotida yang berisi persentase

19
similaritas nukleotida antar pasangan sequence yang dibandingkan tanpa menggunakan
model evolusi apapun.
Analisis dengan Phydit dilakukan dengan membuat file baru dengan membuka menu
File → New atau dengan menekan ikon New Phydit File yang terdapat pada
panel atas bagian paling kiri. Sebuah menu akan muncul dimana kita dapat memasukan
keterangan file. Isi keterangan apabila diperlukan dan apabila sudah, tekan OK. Phydit
kemudian akan menampilkan layar baru bertuliskan No entry to tag. Pada tahap
ini, masukan data melalui Data → Import → GDE (NT Replace) untuk sequence
nukleotida. Pilih data dalam format .gde dan Phydit akan langsung memasukan sequence
berdasarkan entry nama sequence. Penghasilan matriks similaritas nukleotida dapat
dilakukan melalui Analysis → SimTable: Generating Similarity Table
yang terdapat pada panel atas. Matriks similaritas nukleotida terdiri atas dua bagian, yakni
bagian segitiga kanan atas (upper-right triangle) dan segitiga kiri bawah (lower-left
triangle). Phydit akan menanyakan jenis data yang akan dimuat dalam masing-masing
segitiga tersebut. Pada umumnya kita akan memasukan data similaritas nukleotida (NT
Similarity) pada lower-left triangle dan jumlah nukleotida yang berbeda
per total nukleotida yang dibandingkan (NT different/Total Nucleotides)
pada upper-right triangle. Tekan OK dan akan muncul menu Options; tekan
OK lagi untuk melanjutkan analisis. Hasil yang keluar berupa matriks yang dituliskan
pada Notepad. Untuk memudahkan pembacaan, copy seluruh tulisan pada Notepad
tersebut (ctrl+A kemudian ctrl+C) ke MS Excel dan kemudian simpan. Sebagai
tambahan, Phydit juga menyediakan pilihan analisis lainnya yang mencakup:
Alignment Reports, menu ini berfungsi untuk menampilkan hasil alignment sequence
nukleotida/asam amino dalam bentuk text yang secara otomatis dibuka oleh Notepad.
Sequence Statistics, menu ini berfungsi untuk menampilkan frekuensi nukleotida dan
frekuensi asam amino dari sequence yang ada. Apabila kita menggunakan sequence
nukleotida, adanya statistik frekuensi asam amino pada hasil diasumsikan bahwa
sequnce yang dimiliki merupakan coding sequence.
3.2.Konstruksi pohon filogeni menggunakan aplikasi MEGA 4.0
Molecular Evolutionary Genetic Analysis 4.0 (MEGA 4.0) merupakan alat
terintegerasi untuk mengolah pemerataan sekuen, menkonstruksi pohon filogenetik,
memperkirakan perbedaan waktu, memperkirakan kecepatan evolusi molekuler,
menkonstruksi sekuen nenek moyang, dan menguji hipotesis evolusioner. Aplikasi
MEGA 4.0 digunakan oleh para ahli biologi di dalam laboratorium untuk
merekonstruksi sejarah evolusioner dari suatu spesies dan sifat selektif yang
dihasilkan dari kekuatan alam membentuk evolusi gen dan spesies.
Langkah-langkah merekonstruksi pohon filogenetik dengan aplikasi MEGA 4.0
lebih singkat daripada dengan cara diatas.

20
Secara garis besar tahapan rekonstruksi pohon filogenetik dengan menggunakan
aplikasi MEGA 4.0 terdiri atas:
1. Mengunduh sekuens nukleotida yang kita kehendaki dari GenBank
2. Sequence alignment dengan ClustalW
3. Rekonstruksi pohon filogenetik
3.2.1. Mengunduh sekuens nukleotida dari GenBank
Pengunduhan dapat dilakukan dari NCBI (http://www.ncbi.nlm.nih.gov/Genbank)
dengan format FASTA. Diupayakan sekuens nukelotida yang kita gunakan berasal
dari jenis yang sama dan banyaknya sekuen nukleotida tidak jauh berbeda. Apabila
kita memilih sekuen dengan panjang yang perbedaan terlalu jauh maka akan
mengganggu proses alignment dengan ClustalW. Setelah mengunduh format FASTA
dari GenBank kemudian disalin pada program Notepad. Contoh format FASTA
sekuen nukleotida dari B. mycoides GMA030 adalah sebagai berikut:
>gi|398650471|dbj|AB738786.1| Bacillus mycoides gene for 16S rRNA, partial sequence, strain:
GMA030 GATGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGAACCGATTAAGAGCTTGCTCTTAAGAAGT
TAGCGGCGGACGGGTGAGTAACACGTAGGTAACCTGCCTATAAGACTGGGATAACTCCGGGAAACCGGGG
CTAATACCGGATAACATTTTGCACCGCATGGTGCGAAATTGAAAGGCGGCTTCGGCTGTCACTTATAGAT
GGACCTGCGGCGCATTAGCTAGTTGGTGAGGTAACGGCTCACCAAGGCGACGATGCGTAGCCGACCTGAG
AGGGTGATCGGCCACACTGGGACTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTC
CGCAATGGACGAAAGTCTGACGGAGCAACGCCGCGTGAGCGATGAAGGCCTTCGGGTCGTAAAGCTCTGT
TGTTAGGGAAGAACAAGTGCTAGTTGAATAAGCTGGCACCTTGACGGTACCTAACCAGAAAGCCACGGCT
AACTACGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTATCCGGAATTATTGGGCGTAAAGCGC
GCGCAGGTGGTTTCTTAAGTCTGATGTGAAAGCCCACGGCTCAACCGTGGAGGGTCATTGGAAACTGGGA
GACTTGAGTGCAGAAGAGGAAAGTGGAATTCCATGTGTAGCGGTGAAATGCGTAGAGATATGGAGGAACA
CCAGTGGCGAAGGCGACTTTCTGGTCTGCAACTGACACTGAGGCGCGAAAGCGTGGGGAGCAAACAGGAT
TAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAAGTGTTAGAGGGTTTCCGCCCTTTAGTGCT
GAAGTTAACGCATTAAGCACTCCGCCTGGGGAGTACGGCCGCAAGGCTGAAACTCAAAGGAATTGACGGG
GGCCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACAT
CCTCTGACAACCCTAGAGATAGGGCTTCCCCTTCGGGGGCAGAGTGACAGGTGGTGCATGGTTGTCGTCA
GCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCATCATTAA
GTTGGGCACTCTAAGGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCC
CCTTATGACCTGGGCTACACACGTGCTACAATGGACGGTACAAAGAGTCGCAAGACCGCGAGGTGGAGCT
AATCTCATAAAACCGTTCTCAGTTCGGATTGTAGGCTGCAACTCGCCTACATGAAGCTGGAATCGCTAGT
AATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTCACACCACGAGA
GTTTGTAACACCCGAAGTCGGTGGGGTAACCTTTTGGAGCCAGCCGCCTAAGGTGGGACAGATGATTGGG
GTGAAGTCGTAACAAGGTAGCCGTATCGGAA
1. Sequence alignment dengan ClustalW
- Buka aplikasi MEGA 4.0, pilih menu Alignment kemudian beri tanda pada pilihan
Create new alignment dan klik OK.
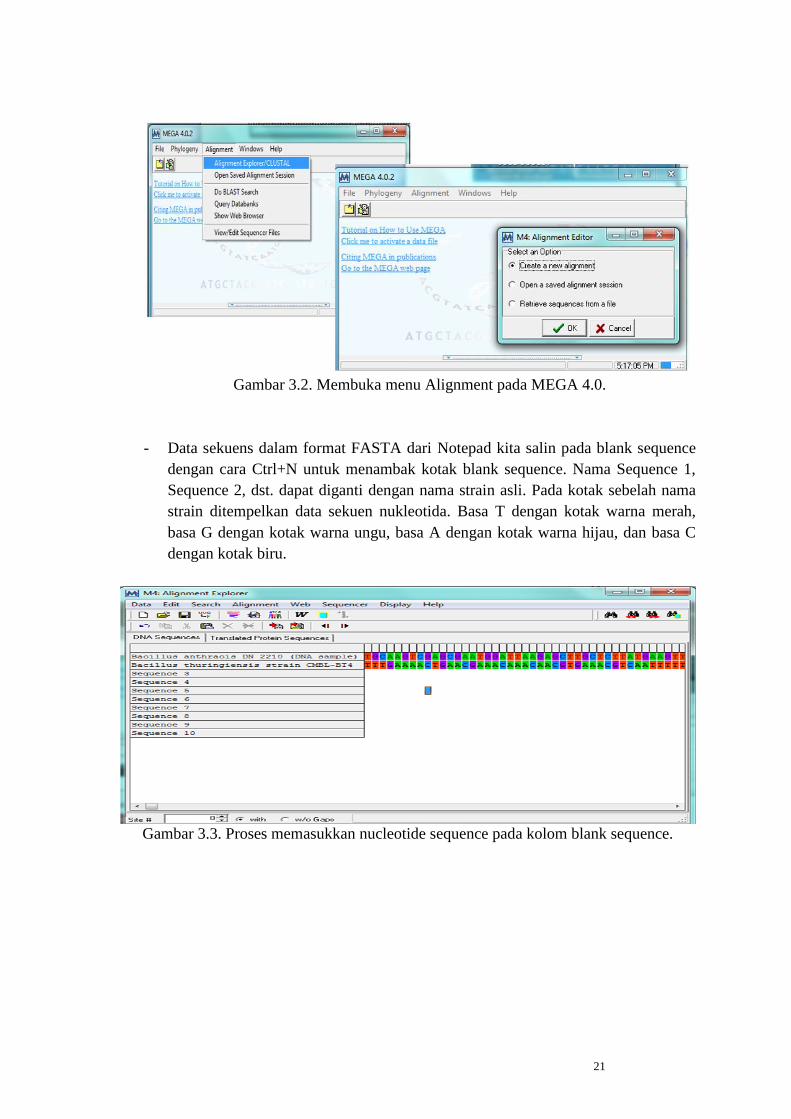
21
Gambar 3.2. Membuka menu Alignment pada MEGA 4.0.
- Data sekuens dalam format FASTA dari Notepad kita salin pada blank sequence
dengan cara Ctrl+N untuk menambak kotak blank sequence. Nama Sequence 1,
Sequence 2, dst. dapat diganti dengan nama strain asli. Pada kotak sebelah nama
strain ditempelkan data sekuen nukleotida. Basa T dengan kotak warna merah,
basa G dengan kotak warna ungu, basa A dengan kotak warna hijau, dan basa C
dengan kotak biru.
Gambar 3.3. Proses memasukkan nucleotide sequence pada kolom blank sequence.
22
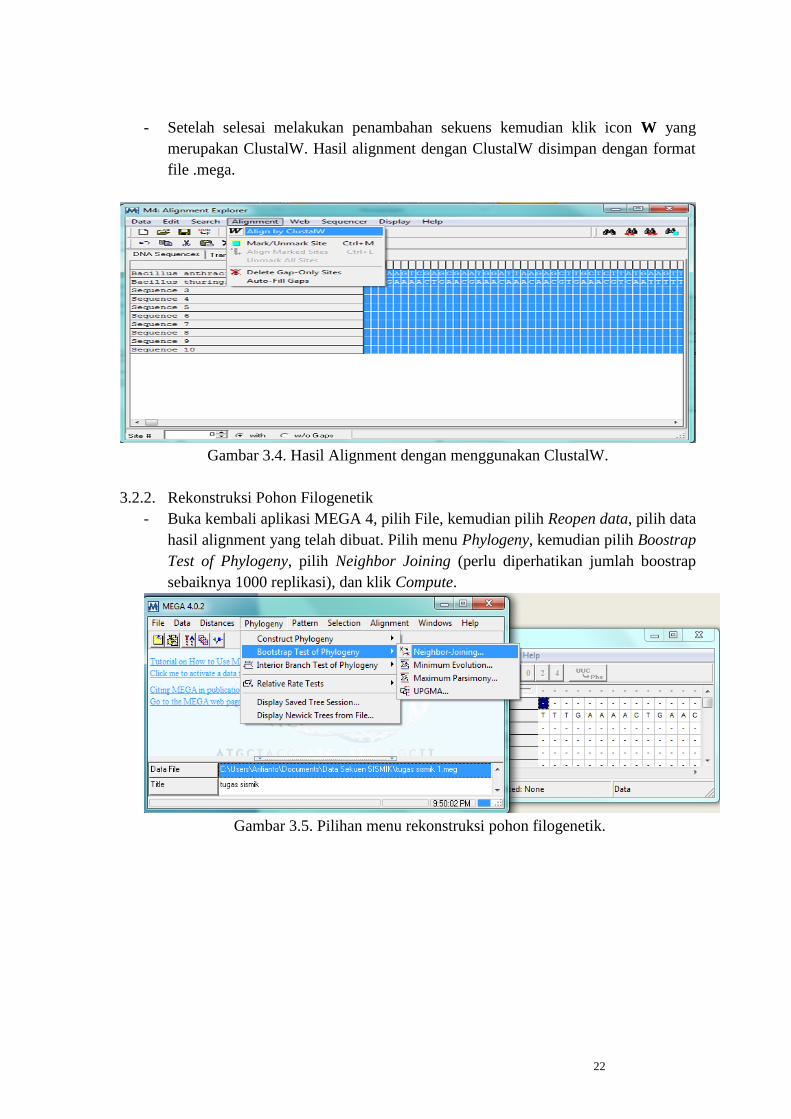
- Setelah selesai melakukan penambahan sekuens kemudian klik icon W yang
merupakan ClustalW. Hasil alignment dengan ClustalW disimpan dengan format
file .mega.
Gambar 3.4. Hasil Alignment dengan menggunakan ClustalW.
3.2.2. Rekonstruksi Pohon Filogenetik
- Buka kembali aplikasi MEGA 4, pilih File, kemudian pilih Reopen data, pilih data
hasil alignment yang telah dibuat. Pilih menu Phylogeny, kemudian pilih Boostrap
Test of Phylogeny, pilih Neighbor Joining (perlu diperhatikan jumlah boostrap
sebaiknya 1000 replikasi), dan klik Compute.
Gambar 3.5. Pilihan menu rekonstruksi pohon filogenetik.
23
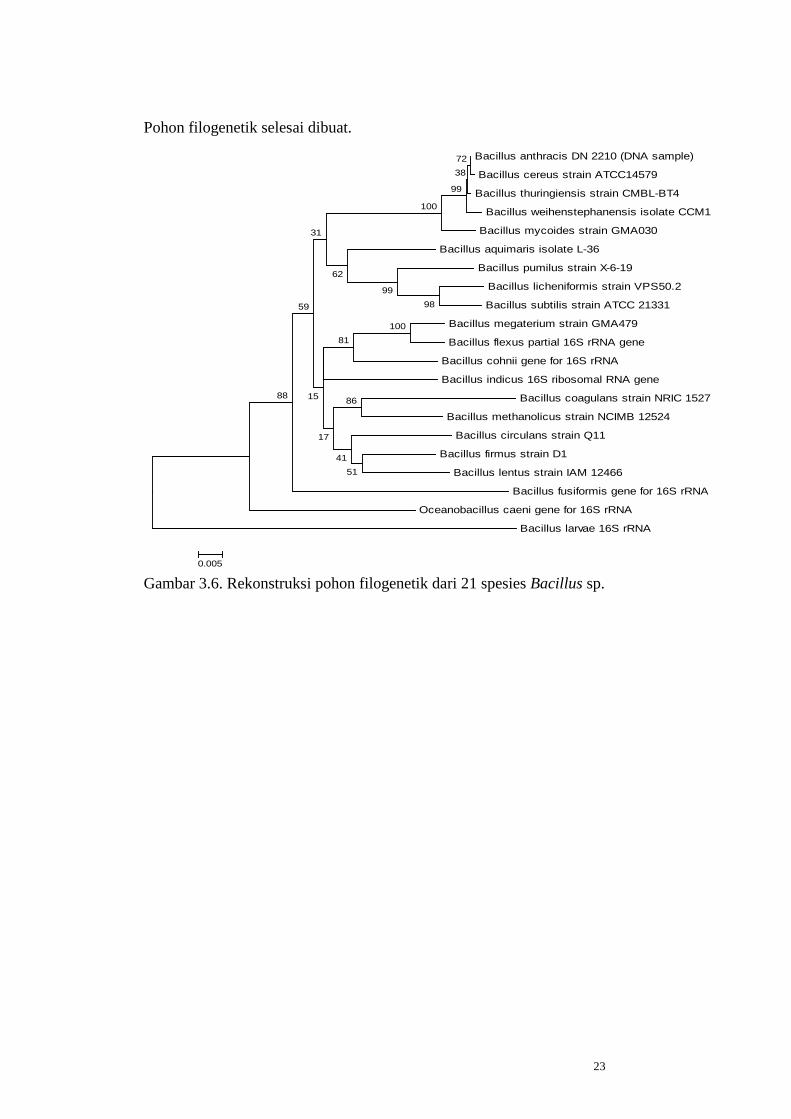
Pohon filogenetik selesai dibuat.
Bacillus anthracis DN 2210 (DNA sample)
Bacillus cereus strain ATCC14579
Bacillus thuringiensis strain CMBL-BT4
Bacillus weihenstephanensis isolate CCM1
Bacillus mycoides strain GMA030
Bacillus aquimaris isolate L-36
Bacillus pumilus strain X-6-19
Bacillus licheniformis strain VPS50.2
Bacillus subtilis strain ATCC 21331
Bacillus megaterium strain GMA479
Bacillus flexus partial 16S rRNA gene
Bacillus cohnii gene for 16S rRNA
Bacillus indicus 16S ribosomal RNA gene
Bacillus coagulans strain NRIC 1527
Bacillus methanolicus strain NCIMB 12524
Bacillus circulans strain Q11
Bacillus firmus strain D1
Bacillus lentus strain IAM 12466
Bacillus fusiformis gene for 16S rRNA
Oceanobacillus caeni gene for 16S rRNA
Bacillus larvae 16S rRNA
72
38
99
100
100
98
99
81
62
88
51
41
31
59
15
17
86
0.005 Gambar 3.6. Rekonstruksi pohon filogenetik dari 21 spesies Bacillus sp.

24
DAFTAR PUSTAKA
Brenner, D. J., N. R. Krieg, J. T. Staley & G. M. Garrity. 2005. Bergey's manual of
systematic bacteriology 2nd
edition, volume 2: The Proteobacteria, Part B: The
Gammaproteobacteria. Springer Pub.: USA.
Chun, J. 1995. Computer-assisted clasification and identification of actinomycetes. Ph.D.
Thesis. University of Newcastle, UK.
Felsenstein, J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by
the author. Department of Genome Sciences, University of Washington, Seattle,
USA.
Goodfellow, M. 1997. Microbial Systematics. In Microbiology and Medical
Microbiology. University of Newcastle Upon Tyne.
Kovach, W.L., 2007. MVSP - A MultiVariate Statistical Package for Windows, ver. 3.1.
Kovach Computing Services, Pentraeth, Wales, U.K.
Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H.
McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T.
J. Gibson, & D. G. Higgins.2007. Clustal W and Clustal X version 2.0.
Bioinformatics 23: 2947–2948.
Page, R. D. M. 1996. TREEVIEW: An application to display phylogenetic trees on
personal computers. Computer Applications in the Biosciences 12: 357–358.
Priest, F. & B. Austin. 1993. Modern Bacterial Taxonomy 2nd
Edition. Chapman & Hall
Pub.: England.
Sembiring, L. 2002. Sistematika Mikrobia (BIO 668) Petunjuk Praktikum. Fakultas
Biologi, Universitas Gadjah Mada, Yogyakarta, Indonesia.
Sembiring, L. 2002. Sistematika Molekular (BIO 765) Petunjuk Praktikum. Fakultas
Biologi, Universitas Gadjah Mada, Yogyakarta, Indonesia.
West, P. A., P. R. Brayton, T. N. Bryant, & R. R. Colwell. 1986. Numerical taxonomy of
vibrios isolated from aquatic environments. International Journal of Systematic
Bacteriology 36 (4): 531–543.






























